The density functional theory (DFT) has established itself as the primary tool to calculate the electronic structure of materials.
At its heart, the method minimizes the Helmholtz free energy using the variational principle. The Helmholtz energy of a system is a functional of the density, and minimizing it gives us the equilibrium density.
DFT for electronic structure calculation takes the eigenvalue of the Schrödinger equation as a functional of the charge density.
Schrödinger’s Equation: Revisited for Many-Particle Systems
The Schrödinger equation is the governing equation for quantum mechanical systems: \begin{equation} \left[-\frac{\hbar^2}{2m}\nabla^2+V(r)\right]\Psi(r)=E\Psi(r) \end{equation}
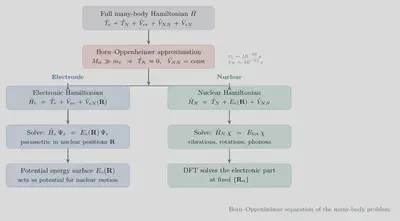
The problem grows intractable when many particles are present. The many-body Hamiltonian for $N$ nuclei and $n$ electrons is:
$$ \begin{align*} \hat{H} =&-\frac{\hbar^2}{2m}\sum_{i}^n\nabla^2_i+ \frac{1}{2}\sum_{i\neq j}\frac{1}{|\mathbf{r}_i-\mathbf{r}_j|} & \qquad \mbox{Electron} \newline & -\frac{\hbar^2}{2M_j}\sum_j^N\nabla^2_j + \frac{1}{2}\sum_{i\neq j}\frac{Z_iZ_j}{|{R_i-R_j}|} & \mbox{Nuclei}\newline & -\sum_{i,j}\frac{Z_j}{|\mathbf{r}_i-\mathbf{R}_j|} & \mbox{Electron-Nucleus} \end{align*} $$The time-independent Hamiltonian $\hat{H}=H(p,q)$ is a function of the generalised momenta and coordinates $(p,q)$. Solving it becomes increasingly difficult for systems with more than one electron.
Even for a simple atom like oxygen, with 8 electrons, and taking a coarse grid of just 10 points in each spatial dimension, we need $10^{24}$ data points. On modern computers, each real number requires 64 bits of memory. To store the wavefunction of a single oxygen atom we would need $64\times 10^{24}$ bits $\approx 8\times 10^{24}$ bytes $\approx 10^{17}$ MB. (To appreciate how large this is, consider that the age of the universe is roughly $4\times 10^{17}$ seconds — the numbers are comparable!)
So we need systematic approximations to solve the problem.
Born–Oppenheimer Approximation
Because protons are roughly $10^3$ times heavier than electrons, they move far more slowly under the same force. This motivates the Born–Oppenheimer (BO) approximation: we freeze the nuclear degrees of freedom and solve only for the electronic structure at fixed nuclear positions.
Under this approximation:
- The nuclear kinetic energy term vanishes: $\frac{\hbar^2}{2M_j}\sum_j^N\nabla^2_j = 0$.
- The nucleus–nucleus repulsion $\frac{1}{2}\sum_{i\neq j}\frac{Z_iZ_j}{|{R_i-R_j}|}$ becomes a constant that shifts the total energy but does not affect the electronic wavefunction.
- The electron–nucleus attraction $\sum_{i,j}\frac{Z_j}{|\mathbf{r}_i-\mathbf{R}_j|}$ depends only on the electronic coordinates $r$ (nuclei are fixed parameters).
The resulting electronic Hamiltonian is: \begin{align*} \hat{H} =&-\frac{\hbar^2}{2m}\sum_{i}^n\nabla^2_i & \mbox{Kinetic energy of electrons}\newline &+ \frac{1}{2}\sum_{i\neq j}\frac{1}{|\mathbf{r}_i-\mathbf{r}_j|} &\mbox{Electron–electron repulsion}\newline & +v_0(r) & \mbox{Electron–nucleus attraction} \end{align*}
The electron–electron repulsion term is still a many-body interaction — handling it requires further approximation.
Mean-Field Approximations
Hartree Approximation
After invoking the Born–Oppenheimer approximation, the remaining challenge is the electron–electron repulsion $\frac{1}{2}\sum_{i\neq j}\frac{1}{|\mathbf{r}_i-\mathbf{r}_j|}$. The simplest treatment sets this to zero (non-interacting electrons), but this gives grossly incorrect results for most properties.
A better strategy is to assume that each electron does not interact with individual others, but instead moves in the mean (average) field generated by all the other electrons combined. This is called the Hartree approximation.
In this picture, the electron–electron repulsion felt by electron $i$ is replaced by the electrostatic potential of the continuous charge density $\rho(r’)$ of all other electrons: \begin{equation} \frac{1}{2}\sum_{i\neq j}\frac{1}{|\mathbf{r}_i-\mathbf{r}_j|} \approx \int \frac{\rho(r’)}{|\mathbf{r}_i-\mathbf{r}’|},dr’ \equiv U_H \end{equation} where $\rho(r’) = \sum_j |\psi_j(r’)|^2$ is the electron number density at position $r’$.
The approximation neglects explicit electron–electron correlations beyond the average field, but captures the dominant Coulomb screening effect, making it a significant improvement over the non-interacting picture.
The full Hartree Hamiltonian then reads: \begin{align*} \hat{H}=-\frac{\hbar^2}{2m}\sum_{i}^n\nabla^2_i + \int \frac{\rho(r’)}{|\mathbf{r}_i-\mathbf{r}’|},dr’ +v_0(r) \end{align*}
Hartree–Fock Approximation
The Hartree approximation is incomplete because it ignores the fact that electrons are fermions: identical spin-$\frac{1}{2}$ particles that obey the Pauli exclusion principle, which requires the many-body wavefunction to be antisymmetric under the exchange of any two electrons.
Fock incorporated this antisymmetry into the Hartree scheme, yielding the Hartree–Fock (HF) approximation. The wavefunction is constructed as a Slater determinant, which automatically satisfies antisymmetry:
$$ \Psi(r_1,r_2\cdots r_N)= \frac{1}{\sqrt{N!}}\begin{vmatrix} \psi_1(r_1) &\cdots &\psi_1(r_N)\\ \vdots & \ddots & \vdots\\ \psi_N(r_1) &\cdots &\psi_N(r_N)\\ \end{vmatrix} $$Swapping any two columns (i.e., exchanging two electrons) changes the sign of the determinant, ensuring the required antisymmetry. The single-particle orbitals $\psi_i$ are then determined by the variational condition:
$$ \frac{\delta}{\delta \psi_j^*(r)}\left[\langle\Psi|\hat{H}|\Psi\rangle-\sum_i^N\epsilon_i\int d^3r |\psi_i(r)|^2\right]=0 $$This procedure introduces an exchange energy term — a purely quantum mechanical contribution absent in the Hartree scheme — which lowers the total energy by keeping electrons of the same spin spatially separated. The correlation energy (everything beyond HF) remains unaccounted for and is the key quantity that DFT’s exchange-correlation functional $E_{\rm xc}$ aims to capture.
Example: Hartree–Fock Calculation for the Helium Atom
The helium atom (two electrons, nucleus with charge $Z=2$) is the simplest system where many-body effects matter, making it an ideal test case.
Hamiltonian: $$ \hat{H} = -\frac{\hbar^2}{2m} (\nabla_1^2 + \nabla_2^2) - \frac{2e^2}{r_1} - \frac{2e^2}{r_2} + \frac{e^2}{|\mathbf{r}_1 - \mathbf{r}_2|} $$
Hartree approximation: Each electron feels the nuclear attraction plus the average repulsion from the other electron. The total wavefunction is a simple product of single-electron orbitals: $\Psi(r_1, r_2) = \psi(r_1)\psi(r_2)$, which is not antisymmetric.
Hartree–Fock improvement: The wavefunction is antisymmetrized via a Slater determinant: $$ \Psi_{HF}(r_1, r_2) = \frac{1}{\sqrt{2}} [\psi_a(r_1)\psi_b(r_2) - \psi_a(r_2)\psi_b(r_1)] $$ For the ground state, both electrons occupy the $1s$ orbital with opposite spins.
Physical consequences:
- The Hartree method overestimates the energy because it ignores exchange.
- The Hartree–Fock method lowers the energy by including exchange, but still neglects electron correlation (all effects beyond a single Slater determinant).
Numerical comparison:
| Method | Ground-state energy (eV) |
|---|---|
| Hartree | > $-77.5$ eV |
| Hartree–Fock | $-77.5$ eV |
| Experiment | $-78.98$ eV |
The gap between Hartree–Fock ($-77.5$ eV) and experiment ($-78.98$ eV) is called the correlation energy. Though small in absolute terms ($\approx 1.5$ eV), it is chemically significant — chemical bond energies are typically of this magnitude. Capturing this missing correlation is the central motivation for DFT and its exchange-correlation functionals.